Gene function research has always been the core of biological research, and establishing a suitable genetic screening system is the best method for gene function research. The earliest radiation and chemical mutagenesis are classical forward genetics methods, which screen out random mutations in the genome based on phenotype and find the connection between phenotype and gene. but the workload of phenotypic screening is huge, and tedious hybridization experiments are needed to determine the location and sequence of genes.
Compared with the forward genetics method, the reverse genetics method is more direct and screens the corresponding phenotype based on the determined gene sequence. In the past, the most commonly used reverse genetic screening methods in biological research were cDNA expression libraries and shRNA libraries, which used gene overexpression and knockdown for phenotypic screening, respectively. The cost of artificially constructed full-gene cDNA expression library is very high. Due to the inherent defects of RNAi technology, shRNA often produces false positives and other abnormal results, which leads to the screening of wrong genes.
The emergence of CRISPR technology has given researchers the ability to manipulate genes in DNA sequences. Compared with the previous generations of gene editing technology, the biggest advantage of CRISPR is that the construction cost is extremely low. When targeting different genes, only the 20 bases of the 5'end of sgRNA need to be changed. With the current nucleic acid chip synthesis technology, it can synthesize millions of sgRNA sequences at the same time to construct an sgRNA library covering the entire gene. The powerful expansion capabilities of the CRISPR system enable CRISPR technology to simultaneously knock out, knock down and activate genes, and establish a comprehensive gene screening system.
Lifeasible has been deeply involved in the field of plant gene editing for many years and has a wealth of technical reserves. We have a rigorous scientific team that can provide excellent end-to-end services and one-stop solutions. We hope that our CRISPR mutation library construction technology can help customers accelerate experiments With the progress of the project, provide customers with satisfactory experimental results.
Using the frameshift knockout method, a single sgRNA can complete the mutation knockout operation. Each gene is designed with 4 to 5 sgRNAs. It takes about 100,000 sgRNAs to cover the entire gene. Nucleic acid chip synthesis technology can easily complete it. After a large number of single-stranded nucleic acids synthesized by the chip, They are converted into double-stranded by PCR, molecular cloning methods such as Gibson reaction can be used to construct a lentiviral vector library of sgRNA. Control the titer of the lentivirus packaged in the library to about 0.3, so that most infected cells contain only one copy of the virus, ensuring the direct correlation between cell phenotype and single gene knockout.
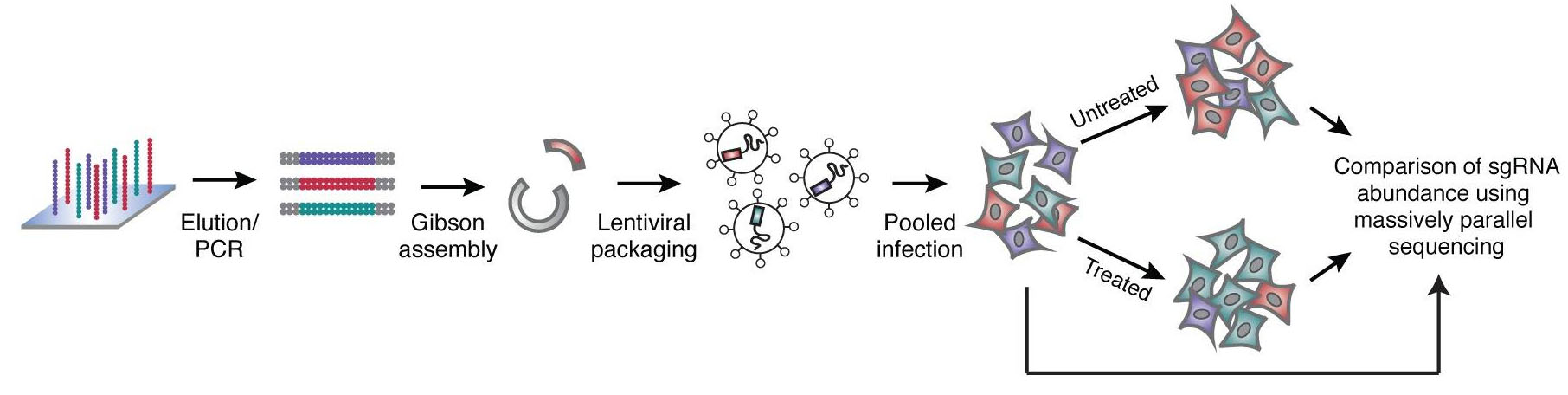 Fig 1. Nucleic acid chip technology construction process of sgRNA library (Wang, et al. 2014)
Fig 1. Nucleic acid chip technology construction process of sgRNA library (Wang, et al. 2014)
Compared with the traditional shRNA knockdown library, the gene knockout library constructed by CRISPR/Cas9 has obvious advantages when the construction cost is basically the same. First of all, gene knockout caused by CRISPR is stable and irreversible at the single-cell level, and gene expression is completely knocked out at the protein level, with good uniformity, which can establish a more direct connection between genes and phenotypes; while shRNA is just Knock down the mRNA level of a gene. The difference in the knock down level has a great influence on the phenotype, resulting in unstable screening results.
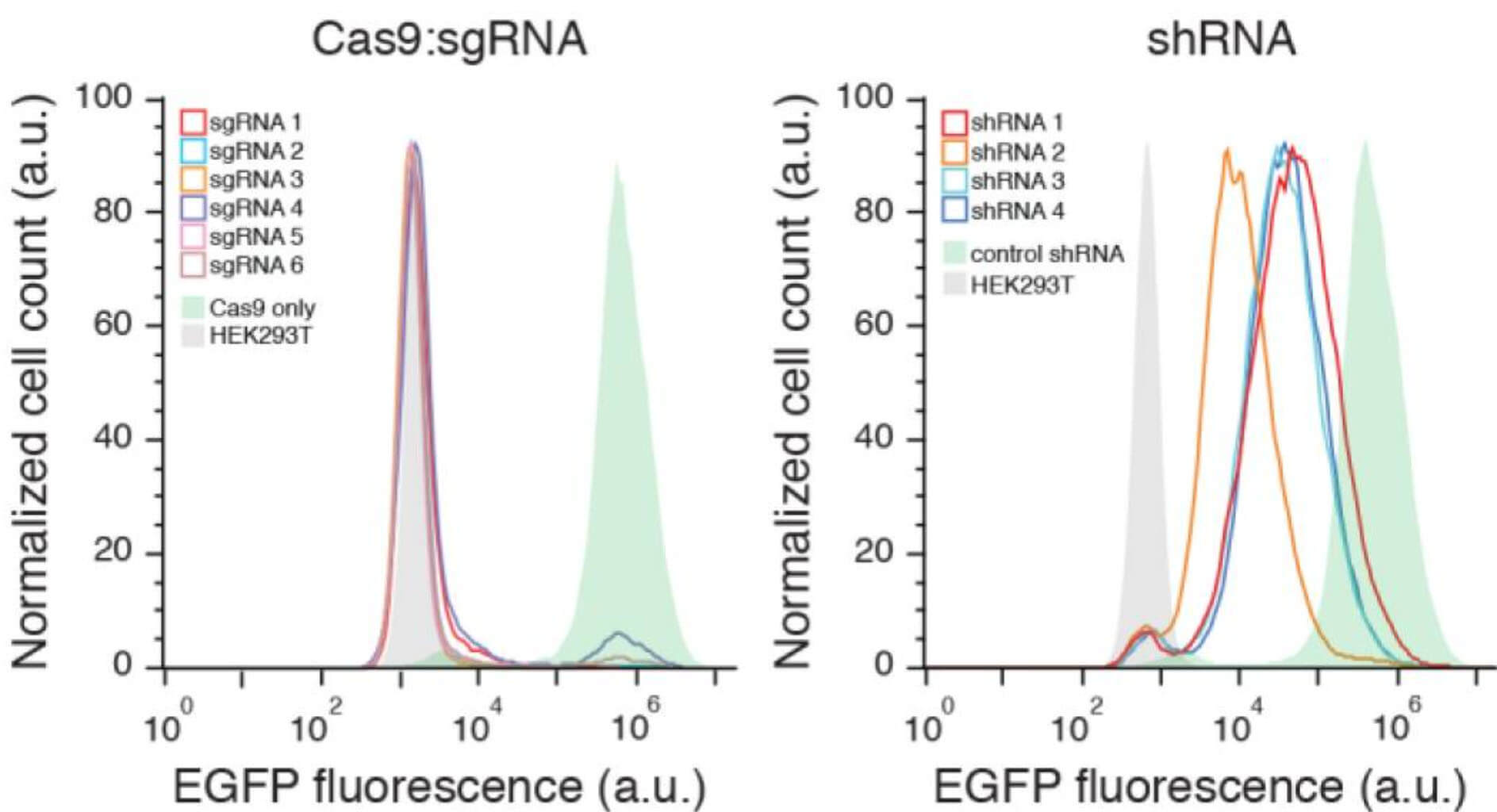 Fig 2. CRISPR/Cas knockout libraries are more stable than shRNA libraries (Shalem, et al. 2014)
Fig 2. CRISPR/Cas knockout libraries are more stable than shRNA libraries (Shalem, et al. 2014)
The CRISPR system has powerful expansion capabilities. CRISPRi is a gene knockdown technology that can replace RNAi technology, and its knockdown ability and accuracy are better than RNAi. Take the screening of non-coding genes as an example. Due to the structural complexity of non-coding RNA, RNAi often cannot knock down non-coding RNA effectively, and frameshift mutations caused by CRISPR knock-out libraries cannot knock down non-coding genes, so CRISPRi knockdown Libraries are another simpler solution. Although the annotations of non-coding genes are generally not complete, the transcription start position of non-coding genes can be roughly determined based on multiple databases, by designing a single sgRNA in a relatively broad region (1kbp) near the transcription start site, the non-coding gene can be knocked down. Using CRISPRi technology, large-scale screening of non-coding genes can also be easily completed (Figure 3).
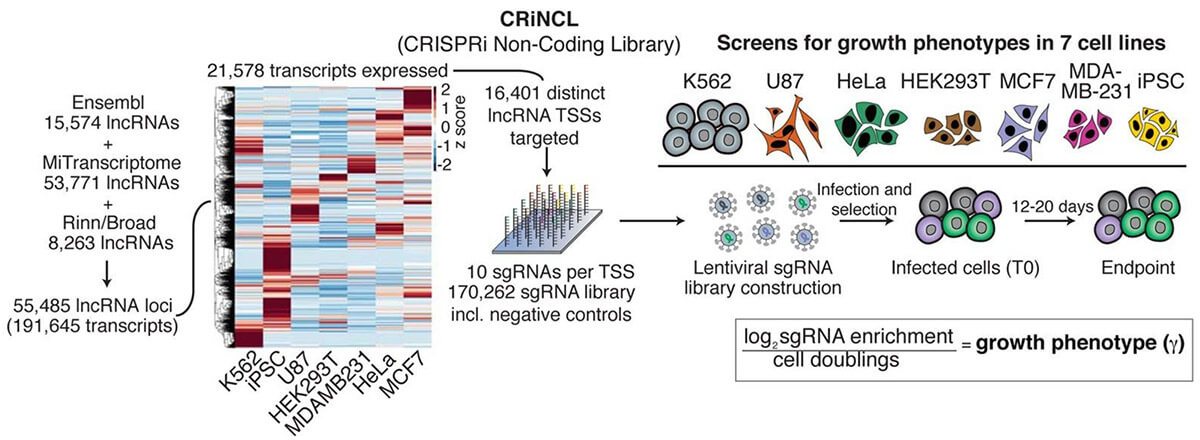 Fig 3. Screening of non-coding genes with CRISPRi gene knockdown library (Liu, et al. 2017)
Fig 3. Screening of non-coding genes with CRISPRi gene knockdown library (Liu, et al. 2017)
There have been many methods for large-scale knockdown or knock-out of genes for genetic screening, and there has been a lack of suitable library screening schemes for activating genes. Traditional cDNA library is through ectopic overexpression to achieve gene activation screening, but the cost of constructing large-scale gene cDNA library is very high, and genes often have multiple forms of splicing, cDNA library may only choose one of them and ignore other forms. The emergence of CRISPRa technology has given us the tools to activate gene expression in situ. Although several mainstream CRISPRa technologies, such as VPR, SAM and Suntag, have more complex compositions, they did not increase the cost of sgRNA construction, CRISPRa gene activation libraries can still be obtained by constructing sgRNA libraries. (Figure 4). The emergence of the CRISPRa gene activation library fills the gaps in GOF (gain-of-function) in gene screening. Together with the LOF (loss-of-function) screening method composed of CRISPR and CRISPRi libraries, it provides a scientific community with perfect Solutions for gene function screening and research.
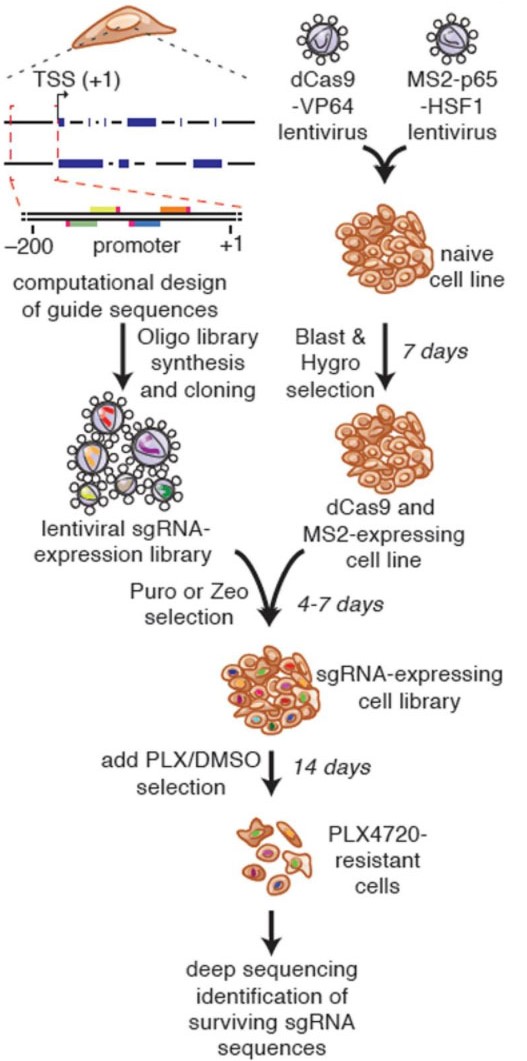 Fig 4. Process of full gene screening using CRISPRa SAM technology (Konermann, et al. 2015)
Fig 4. Process of full gene screening using CRISPRa SAM technology (Konermann, et al. 2015)
By combining CRISPR screening technology and single-cell sequencing, single-gene changes and single-cell transcriptome changes are established. We can cover whole-gene CRISPR editing through a large number of single-cell sequencing, and obtain the connection between each edited gene and the entire transcriptome, which greatly improves the information throughput of the CRISPR screening system. The combination of CRISPR screening system and single-cell sequencing is not complicated. Normal sgRNA is transcribed by PolIII promoter U6 and does not have polyA at the end, so it cannot be detected by single-cell sequencing. The additional use of PolII promoter in library design can transcribe the entire U6-sgRNA sequence into RNA with a polyA tail, which can be detected by single-cell sequencing without interfering with the normal transcription of U6 sgRNA sequence. Based on this design, in the single-cell sequencing results, a single sgRNA sequence can be found in the sequencing results of a single cell, and the connection between the sequencing result and the sgRNA target gene can be established.
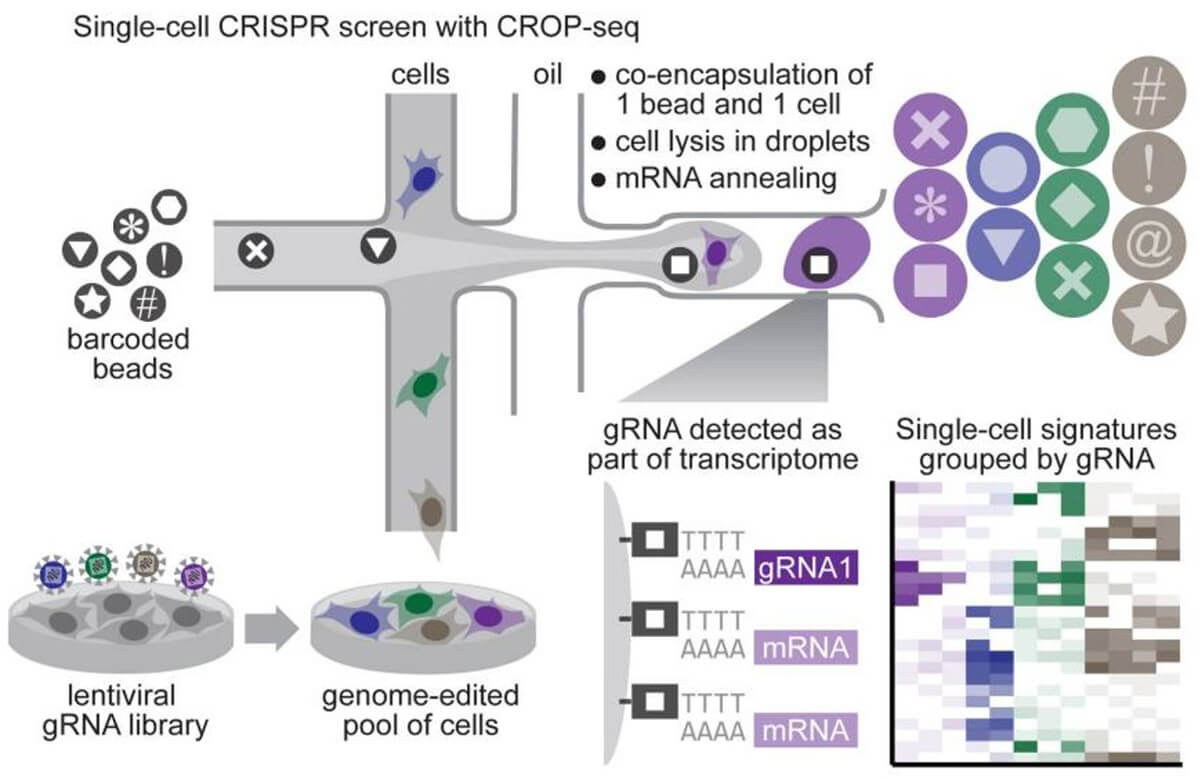 Fig 5. Process of CRISPR screening system based on single cell sequencing (Datlinger et al. 2017)
Fig 5. Process of CRISPR screening system based on single cell sequencing (Datlinger et al. 2017)
Lifeasible can provide our customers with a full range of gene editing technology and related services. We hope that our technology and services can help you achieve new breakthroughs. Inquiries and orders are also welcome. For more information, please contact Lifeasible.
References
Get Latest Lifeasible News and Updates Directly to Your Inbox

